Cistinė fibrozė
Sinonimai plačiąja prasme
Cistinė fibrozė, plaučiai
Anglų kalba: mukoviscidozė, cistinė fibrozė
Cistinės fibrozės apibrėžimas
Cistinė fibrozė yra paveldima liga. Paveldėjimas mediciniškai vadinamas autosominiu recesyviniu. Cistinė fibrozė (cistinė fibrozė) nėra paveldima lytinėse X ir Y chromosomose, bet 7 autosominėje chromosomoje.
Perskaitykite mūsų bendrą straipsnį apie medžiagų apykaitos sutrikimus: Metabolizmo sutrikimai - ką tai reiškia?
Mutacija yra vadinamajame CFTR gene. Recesyvus reiškė, kad ligai išsivystyti turėjo būti dvi sugedusios geno kopijos. Jei žmogus turi sveiką ir mutavusį geno vietą atitinkamoje 7 chromosomoje, liga nepasireiškia.
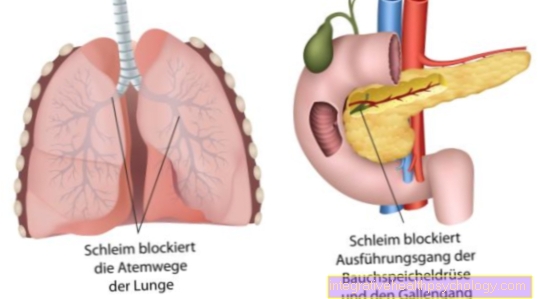
Rezultatas yra patologinis genų produktas. Tokiu būdu koduojama Chlorido kanalai yra sulaužytos. Dėl netinkamų chloridų kanalų susidaro storos gleivės visose egzokrininėse liaukose.
Šios egzokrininės liaukos, t. Y. Liaukos, išskiriančios sekreciją į išorę, apima:
- kasa
- plonoji žarna
- kvėpavimo takų sistema su plaučiais ir bronchų sistema
- tulžies takų ir
- taip pat Prakaito liaukos
Santrauka
Cistinė fibrozė yra Paveldima liga. Paveldima tokiu būdu, kad ji nepriklauso nuo lyties ir tik su du genai su defektais atsiranda. Tai yra dažniausiai pasitaikantis autosominis recesyvinis palikimas.
Pasekmės yra kietos gleivinės formacijos visų egzokrininių liaukų, tokių kaip plaučių, kasos ir prakaito liaukos. Jie tuo remiasi sutrikdytas chlorido pernešimas tarp kameros vidaus ir išorės (skaitykite toliau: Chloridas kraujyje). Įjungtas mutavęs genas 7 chromosoma ir sukelia platų organų dalyvavimą, darantį atitinkamą poveikį kvėpavimui, virškinimui ir reprodukcijai.
Deja, terapija gali tik palengvinti simptomus, bet neišgydyti. Gyvenimo trukmė pacientams, sergantiems cistine fibroze palyginti žemas.
Kadangi tai yra recesyvinė paveldima liga, yra žmonių, kurie nešioja pakeistą geną, tačiau nepatiria pačios ligos. Tokie asmenys vadinami Funkcijų vežėjas arba Laidininkai, t.y., vežėjai. Šie žmonės neturi cistinės fibrozės, nes kita geno kopija yra nepažeista, o sergantis nėra pakankamai stiprus, kad vyrautų.
Tačiau ji gali perduoti šią sugedusią geno kopiją savo palikuonims. Jei modifikuoto geno jau pakaktų ligai sukelti, tai būtų vadinamasis dominuojantis palikimas. Tokį palikimą galima rasti, pavyzdžiui, Chorea Huntingtonas. Daugiau apie šią ligą galite sužinoti mūsų temoje Chorea Huntingtonas.
Apie 1:2500 yra Ligos rodiklis naujagimiams Vokietijoje. Vežėjas yra apie visus 25. Vokietijos gyventojų.
Pagrindinė priežastis

Cistinę fibrozę sukelia 7 chromosomos geno mutacija. Ši chromosoma yra autosominė, o ne lytinė chromosoma.
Kiekvienas turi 44 autosomines chromosomas (dvi identiškos kiekvienos versijos) ir dvi lytines chromosomas. Dėl šios 7 chromosomos mutacijos susidaro trūkūs chlorido kanalai. Chloridas negali absorbuoti (pakartotinai absorbuoti) iš liaukų sekretų, nes receptorius, chlorido jungties taškas, nėra integruotas į liaukų kanalus.
Dėl netinkamos išvaizdos ir struktūros jis atiduodamas kasybai. Sutrinka natūralus chlorido keitimasis tam tikrais chlorido kanalais. Šiuos vadinamuosius kanalus sudaro baltymai. Mūsų DNR yra užkoduota daugybė baltymų. Dėl genetinio chlorido kanalų defekto dehidratuotos ir sunkios gleivės gamina iš visų liaukų, kurios išskiria jų sekreciją į išorę. Tuomet gleivės iš dalies blokuoja ortakius ar kvėpavimo takus plaučiuose.
Taip pat skaitykite apie tai Chromosomų mutacija
Cistinės fibrozės diagnozė

Tipiški simptomai, prasidedantys kūdikystėje, yra novatoriški diagnozuojant cistinę fibrozę.
Šį įtarimą sustiprina teigiama šeimos istorija (tėvo / motinos ar artimų giminaičių liga). Teigiama šeimos istorija reiškia, kad šeimoje yra arba jau buvo cistinės fibrozės atvejų - iš motinos ar tėvo pusės.
Kasos fermentų trūkumą galima nustatyti ir išmatose. Bet kokius kvėpavimo takų užsikimšimus galima aptikti rentgeno būdu atliekant krūtinę.
Cistinės fibrozės diagnozei taip pat padeda prakaito tyrimas, kurio metu matuojamas chloro kiekis prakaite. Jei viršijama tam tikra vertė ir taikomi ir kiti simptomai, diagnozė yra gana fiksuota. Dažnai tėvai patys pastebi padidėjusį druskos kiekį kūdikio prakaite.
Neišnešiotas vaikas taip pat gali būti tiriamas dėl šios paveldimos ligos. Naudojant amniono skysčio punkciją (Amniocentezė) vaisiaus ląstelės pašalinamos ir tiriamas mutavęs genas.
Skaitykite daugiau šia tema: Vaiko rentgeno tyrimas
Cistinės fibrozės gydymas
Kiekvienas, kurį paveikė cistinė fibrozė, gaus patarimą viename Cistinė fibrozė - ambulatorinis skyrius arba patarimai iš Žmogaus genetikas (Paveldimų ligų specialistas). Tai gali padėti pagerinti gyvenimo kokybę arba, jei norite turėti vaikų, apskaičiuoti sergančio vaiko tikimybę. Jei tėvai yra derlingi ir derlingi.
Priešingu atveju gydymas yra simptominis, nes priežasties - geno trūkumo - nepavyksta pašalinti.
Nepagydoma liga
Cistinė fibrozė (cistinė fibrozė) iki šiol yra nepagydoma liga.
Cistinės fibrozės atveju svarbu tinkamai suvartoti valgomosios druskos (Natrio chloridas, NaCl). Žinoma, siekiama gleivinės analizės. Mukolizė yra gleivių ištirpinimas, ypač plaučiuose, kad būtų lengviau kvėpuoti.
Vaistai ir įkvėpus galima palengvinti simptomus. Jei pastebimai pablogėja plaučių funkcija, galima skirti deguonies.
Atliekant intensyvią kineziterapiją (fizioterapija), pavyzdžiui, atliekant masažą ir kvėpavimo pratimus, taip pat gydomi plaučių pokyčiai, kuriuos sukelia cistinė fibrozė.
Dažnai liga baigiasi reikalaujamu plaučių persodinimu. Tačiau laukiančiųjų sąrašai yra ilgi.
Geriamoji kasos fermentų ir riebaluose tirpių vitaminų dozė taip pat yra terapijos dalis. Todėl kasos užduotis turi būti palaikoma, tiksliau, pakeista. Riebaluose tirpūs vitaminai yra A, D, E ir K. Jie turi būti duodami tiesiai į kraują, nes dėl virškinimo fermentų trūkumo negali absorbuotis iš maisto.
Dietoje taip pat turėtų būti daug kalorijų, nes tik dalį jų galima gauti iš maisto.
Kad būtų išvengta papildomų komplikacijų, tokių kaip gripas ar pneumonija, rizikos veiksnių, vaikas turėtų būti paskiepytas. Rekomenduojamos šios vakcinacijos:
- tymai
- Pneumokokai
- gripas
Skaitykite daugiau šia tema: Superinfekcija
Žinoma, dėl šių priemonių būtina pasitarti su gydytoju, su kuriuo reikėtų aptarti riziką.
Šiais laikais didelę viltį dėl cistinės fibrozės terapijos dedame į genetinius tyrimus. Į žmogaus genomą bandoma įvesti trūkstamą genetinę informaciją. Mes ieškome vektorių, kurie galėtų įsisavinti šią užduotį. Vektoriais gali būti, pavyzdžiui, bakterijų arba virusų DNR, sugebantys į mūsų genomą įtraukti sveiką dažnį.
Šiuo metu išbandomas negimusių pacientų terapinis požiūris. Pelėms pelių embrionams jau pavyko įnešti sveiką geną, kuriame buvo teisinga genų seka, per amniocentezę (inniuliacija amniono skysčiu). Taigi šiose pelėse buvo pagamintas sveikas CFTR genas. Amniocentezė yra vaiko ląstelių punkcija ir pašalinimas iš amniono. Tai atliekama per motinos pilvo sieną.
Tačiau Vokietijoje ši intrauterininės (= gimdoje = gimdoje) „terapijos“ forma yra draudžiama.
profilaktika
A prevencinė priemonė šia prasme jis neegzistuoja, nes tai yra paveldima liga.
Tačiau galima aplankyti žmogaus genetinių konsultacijų centrą (paprastai jį rasite universitetinėse ligoninėse). Čia apskaičiuojama, kokia didelė rizika būtų perduoti ligą vaikams.
Šis patarimas visada naudingas, jei šeimoje yra cistinė fibrozė.
Taip pat vienas Prenatalinė diagnostika verta stengtis. Čia prieš gimdymą (t. Y. Prieš gimdymą) a Amniono skysčio tyrimas (Amniocentezė) atliko. Vaisiaus ląstelės (vaiko ląstelės) paimamos iš amniono skysčio ir tiriama mutavusio geno DNR.
Cistinės fibrozės prognozė
Deja, vidutinė gyvenimo trukmė pacientams, sergantiems cistine fibroze, yra tik 32-37 metai. Manoma, kad naujagimių, gimusių šia liga, gyvenimo trukmė yra apie 45–50 metų.
Prognozė labai priklauso nuo terapijos ir nuo jos laikymosi.
Taigi pats pacientas ir jo motyvacija vaidina svarbų vaidmenį.














.jpg)